Mechanisms of key genes responsible for cuproptosis
| PMID | Mechanisms | Date | Gene |
|---|---|---|---|
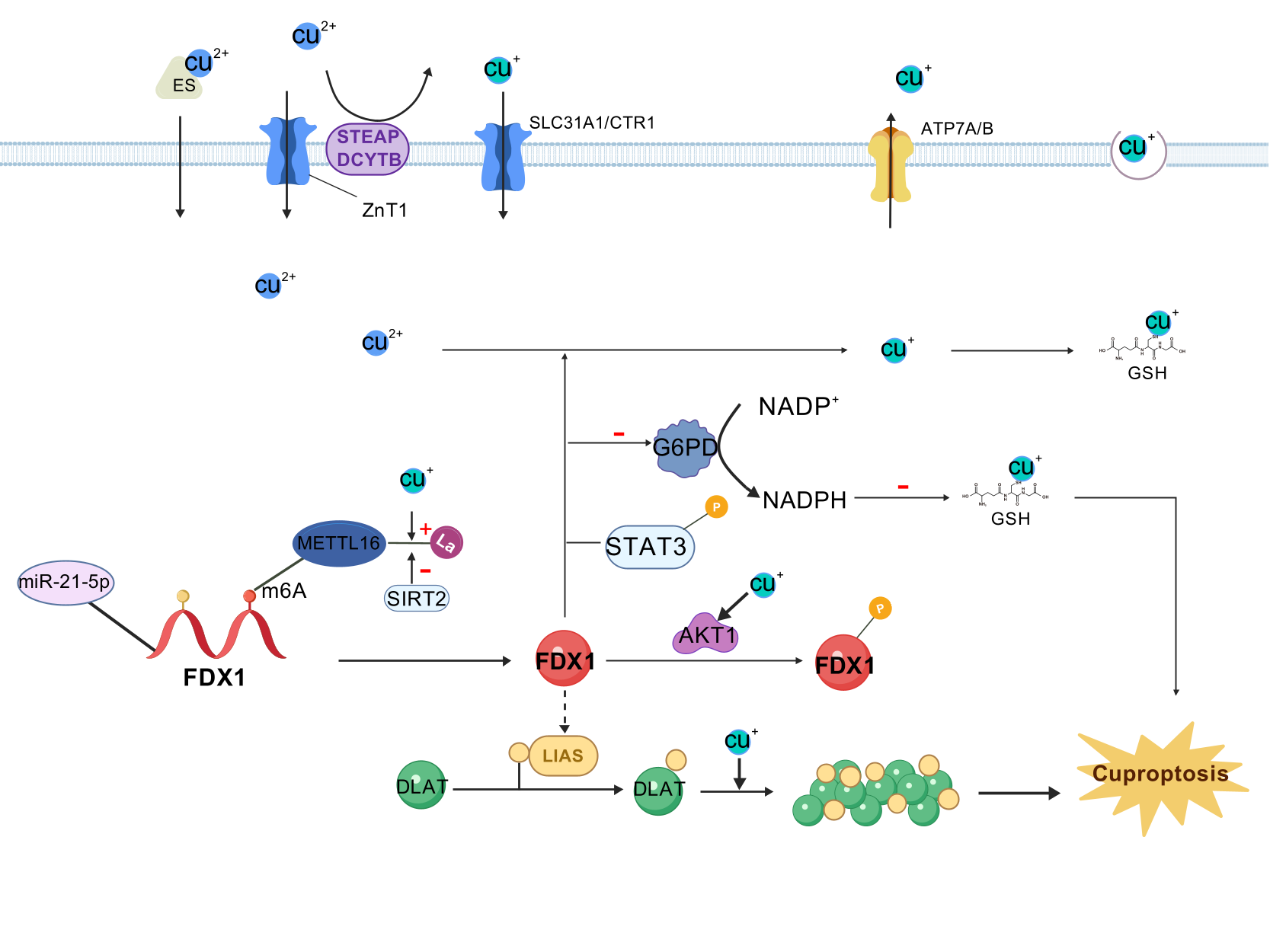
| 35298263 | FDX1 and lipoic acid genes are critical mediators of copper ionophore-induced cell death.FDX1 was considered may act as the upstream of LA pathway, and under the regulation of FDX1, LIAS linked lipoyl moiety to DLAT, which was essential for the function of mitochondrial PDC. Cu(I) directly bound with lipoyl moiety of lipoylated DLAT through disulfide bond and further lead to DLAT oligomerization and further proteotoxic stress and finally result in cell death. | 17-Mar-22 | |
| 37863889 | METTL16, an atypical methyltransferase, is a critical mediator of cuproptosis through the m6A modification on FDX1 mRNA. Furthermore, copper stress promotes METTL16 lactylation at site K229 followed by cuproptosis. The process of METTL16 lactylation is inhibited by SIRT2. Elevated METTL16 lactylation significantly improves the therapeutic efficacy of the copper ionophore- elesclomol. | 20-Oct-23 | |
| 37119432 | FDX1 interacts with G6PD, and reduces its protein stability, which predominantly affects the cellular redox-regulating systems. Then, the reduced G6PD activity enhances cuproptosis via down-regulating NADPH and GSH levels. | 29-Apr-23 | |
| 36611966 | we identified the miR-21-5p/FDX1 axis in ccRCC and experimentally verified that miR-21-5p directly binds the 3'-UTR of FDX1 to mediate its degradation.Our data clearly elucidated that knockdown of FDX1 significantly increased the phosphorylation levels of STAT3 at Y705, indicating that STAT3 signaling may be involved in the FDX1-mediated cuproptosis-independent phenotypic changes in ccRCC cells. | 31-Dec-22 | |
| 35298263 | FDX1 and lipoic acid genes are critical mediators of copper ionophore-induced cell death.FDX1 was considered may act as the upstream of LA pathway, and under the regulation of FDX1, LIAS linked lipoyl moiety to DLAT, which was essential for the function of mitochondrial PDC. Cu(I) directly bound with lipoyl moiety of lipoylated DLAT through disulfide bond and further lead to DLAT oligomerization and further proteotoxic stress and finally result in cell death. | 17-Mar-22 | |
| 37949877 | We validated that elevated MELK expression enhanced the activity of PI3K/mTOR signaling and subsequently promoted Dihydrolipoamide S-Acetyltransferase (DLAT) expression and stabilized mitochondrial function. This regulatory effect helped to improve mitochondrial respiration, eliminate excessive intracellular reactive oxygen species (ROS), reduce intracellular oxidative stress/damage and the possibility of mitochondria-induced cell fate alternations, and ultimately promote the progression of HCC. | 18-Dec-23 | |
| 39460738 | Mechanistic studies showed that DLAT directly interacts with YAP1, leading to the dephosphorylation and activation of YAP1 and its increased nuclear translocation, thereby transcriptionally activating and regulating downstream oncogenes, promoting the malignant phenotype of TNBC. Rescue experiments indicated that DLAT promotes the malignant behavior of TNBC through a YAP1-dependent pathway. | 26-Oct-24 | |
| 35298263 | FDX1 and lipoic acid genes are critical mediators of copper ionophore-induced cell death.FDX1 was considered may act as the upstream of LA pathway, and under the regulation of FDX1, LIAS linked lipoyl moiety to DLAT, which was essential for the function of mitochondrial PDC. Cu(I) directly bound with lipoyl moiety of lipoylated DLAT through disulfide bond and further lead to DLAT oligomerization and further proteotoxic stress and finally result in cell death. | 17-Mar-22 | |
| 39930254 | I/R significantly enhances PCSK9 expression and therefore provokes inflammatory responses, oxidative stress, mitochondrial dysfunction, and cuproptosis by directly interacting with LIAS, which results in cardiac remodeling and dysfunction. | 11-Feb-25 | |
| 40265013 | lnc-CNNM3-DT overexpression downregulated LIAS expression and decreased intracellular copper ion levels. | 8-Apr-25 | |
| 38041690 | We confirmed that LIPT1 downregulates the copper chaperone gene antioxidant 1 (ATOX1), thereby impeding NSCLC progression. | 2-May-23 | |
| 37668796 | Further molecular experiments indicated that LIPT1 silencing repressed the expression of peroxisome proliferator-activated receptor gamma (PPARγ) and inactivated the AKT/GSK-3β/β-catenin signaling axis. | 5-Sep-23 | |
| 39198615 | Mechanistically, YTH N6-Methyladenosine RNA Binding Protein F2 (YTHDF2) promoted the degradation of LIPT1 mRNA in a m6A-dependent manner. | 28-Aug-24 | |
| 40176113 | This study revealed that circKIAA1797 inhibits intracellular cuproptosis by binding to the ferredoxin 1 (FDX1) mRNA, decreasing FDX1 mRNA stability, inhibiting FDX1 expression, and binding to the signal transducer and activator of transcription 1 (STAT1) protein and inhibiting lipoyltransferase 1 (LIPT1) transcription; moreover, circKIAA1797 promotes the closure of the mitochondrial permeability transition pore (mPTP), inhibits cuproptosis, and ultimately promotes lung cancer development. | 2-Apr-25 | |
| 40213908 | The study shows that Ras-related protein Rab-18 (RAB18), a protein involved in lipid metabolism, inhibits lipophagy, upregulates Carnitine palmitoyltransferase 1A (CPT1A), and promotes succinylation of dihydrolipoamide dehydrogenase (DLD) at site K320, triggering cuproptosis in HSCs. | 11-Apr-25 | |
| 37123026 | After knockdown of DLD expression, it was found that DLD regulated metabolic pathways by suppressing the intracellular NAD+/NADH ratio, which then in turn suppressed tumor cell proliferation detected by MTT assay. | 6-Apr-23 |