RNA Modifications 
 Manuals:
Manuals:
Step1: Users can access the RNA modifications query page by clicking on the "RNA Modifications" module of the home page or the options of the navigation bar to query all modification sites of a certain modification type that users are interested in;
Step2: Select the targeted genome and get all RNA modification sites of this species, default is Homo Sapiens (hg38);
Step3: Click the title of the result table to resort the results; Or click "ModID" to display the detail information about this modification site. And the details will be displayed in the detail information section that is below the query page;
Step4: In "Details", there is a specific explanation when the mouse hover the logo in the upper right corner of each entry. The detailed list will be shown if users click the "Show Detail" button.

Fig.1 The workflow of "RNA Modifications" module
 Descriptions:
Descriptions:
♥ Modification sites are classified into the following gene biotypes: protein-coding genes (mRNAs), tRNAs, rRNAs, Mt-tRNAs, lincRNAs, pseudogenes etc.; and gene features including CDS, 3′-UTR, 5′-UTR, intron, exon and intergenic. All types of RNA modification sites are distributed in above-mentioned gene features.
♥ "Seq Type": the value is the type of high-throughput sequencing data like m6A-seq, "na" means the source of this site is a public database like Modomics.
♥ For m6A, m6A sites are distributed on different gene biotypes including protein_coding, miRNA, rRNA, Mt_rRNA, tRNA, Mt_tRNA, ncRNA, scaRNA, scRNA, snoRNA, snRNA, sRNA, lincRNA, lncRNA, Alu, intergenic, IG_C_gene, IG_J_gene, IG_C_pseudogene, IG_V_gene, retained_intron, retained_intronretained_intron, translated_processed_pseudogene, transcribed_unprocessed_pseudogene, pseudogene, transcribed_processed_pseudogene, processed_transcript, misc_RNA and others; the "seqType" of m6A contains m6A-seq, MeRIP-Seq, m6A-CLIP/IP, miCLIP, m6A-REF-seq, DART-seq and MAZTER-seq.
♥ For m1A, m1A sites are distributed on different gene biotypes including protein_coding, miRNA, rRNA, Mt_rRNA, tRNA, Mt_tRNA, snoRNA, snRNA, sRNA, srpRNA, lincRNA, lncRNA, Alu, intergenic, retained_intron, transcribed_unprocessed_pseudogene, pseudogene, processed_transcript, misc_RNA and others; the "seqType" of m1A contains m1A-seq and m1A-MAP.
♥ For m5C, m5C sites are distributed on different gene biotypes including protein_coding, miRNA, rRNA, Mt_rRNA, tRNA, Mt_tRNA, ncRNA, scaRNA, scRNA, snoRNA, snRNA, lincRNA, lncRNA, Alu, intergenic, IG_C_gene, IG_V_gene, retained_intron, transcribed_unprocessed_pseudogene, transcribed_processed_pseudogene, pseudogene, processed_transcript, misc_RNA and others; the "seqType" of m5C contains m5C-RIP, iCLIP-seq and Bisulfite-seq.
♥ For m7G, m7G sites are distributed on different gene biotypes including protein_coding, rRNA, tRNA, lincRNA, lncRNA, Alu, intergenic, retained_intron, processed_pseudogene, transcribed_processed_pseudogene, processed_transcript and others; the "seqType" of m7G contains m7G-seq, BoRed-seq and m7G-RIP-seq.
♥ For 2'-O-Me, Nm sites are distributed on different gene biotypes including protein_coding, miRNA, rRNA, Mt_rRNA, tRNA, ncRNA, scRNA, snoRNA, snRNA, lincRNA, lncRNA, Alu, intergenic, retained_intron, transcribed_unprocessed_pseudogene, pseudogene, transcribed_processed_pseudogene, processed_transcript, misc_RNA and others; the "seqType" of 2'-O-Me contains RiboMeth-seq and Nm-seq.
♥ For pseudouridine, Pseudo sites are distributed on different gene biotypes including protein_coding, miRNA, rRNA, Mt_rRNA, tRNA, Mt_tRNA, ncRNA, scaRNA, snoRNA, snRNA, lincRNA, lncRNA, Alu, intergenic, retained_intron, transcribed_unprocessed_pseudogene, pseudogene, processed_pseudogene, transcribed_processed_pseudogene, processed_transcript, misc_RNA, transposable_element and others; the "seqType" of pseudouridine contains Ψ-seq, PSI-seq, Pseudo-seq and CeU-seq.
♥ For RNA-editing, A-I sites are distributed on different gene biotypes including protein_coding, miRNA, pre_miRNA, rRNA, tRNA, ncRNA, scaRNA, scRNA, snoRNA, snRNA, sRNA, srpRNA, lincRNA, lncRNA, Alu, intergenic, IG_J_gene, IG_J_pseudogene, IG_V_gene, retained_intron, transcribed_unprocessed_pseudogene, pseudogene, processed_pseudogene, transcribed_processed_pseudogene, processed_transcript, misc_RNA and others; RNA-editing data are from 3 public databases, they are DARNED, RADAR, REDIportal and FairBase.
♥ For 2'-O-Me, Nm sites are distributed on different gene biotypes including protein_coding, miRNA, rRNA, tRNA, Mt_tRNA, ncRNA, snoRNA, snRNA, lincRNA, lncRNA, Alu, intergenic, retained_intron, pseudogene, processed_pseudogene, processed_transcript and others; other types of RNA modifications are download from other pubic databases, including Modomics, snOPY, snoRNABase and Yeast-snoRNADataBase.
Mechanism
Manuals:
Step1: Users can access the Mechanisms query page by clicking on the "Mechanism" module of the home page or the options of the navigation bar to query all modification sites that guided by Enzymes and/or snoRNAs users are interested in;
Step2: Select the targeted genome and enzyme/snoRNA users need, then get all RNA modifications guided by enzymes and/or snoRNAs of this species, default is Homo Sapiens (hg38);
Step3: Click the title of the result table to resort the results; Or click "Enzyme ID" or "snoRNA Name" to display the detail information of RNA modifications about this enzyme or snoRNA. And the details will be displayed in the detail information section that is below the query page;
Step4: In "Details", there is a specific explanation when the mouse hover the logo in the upper right corner of each entry. The detailed list will be shown if users click the "Show Detail" button.
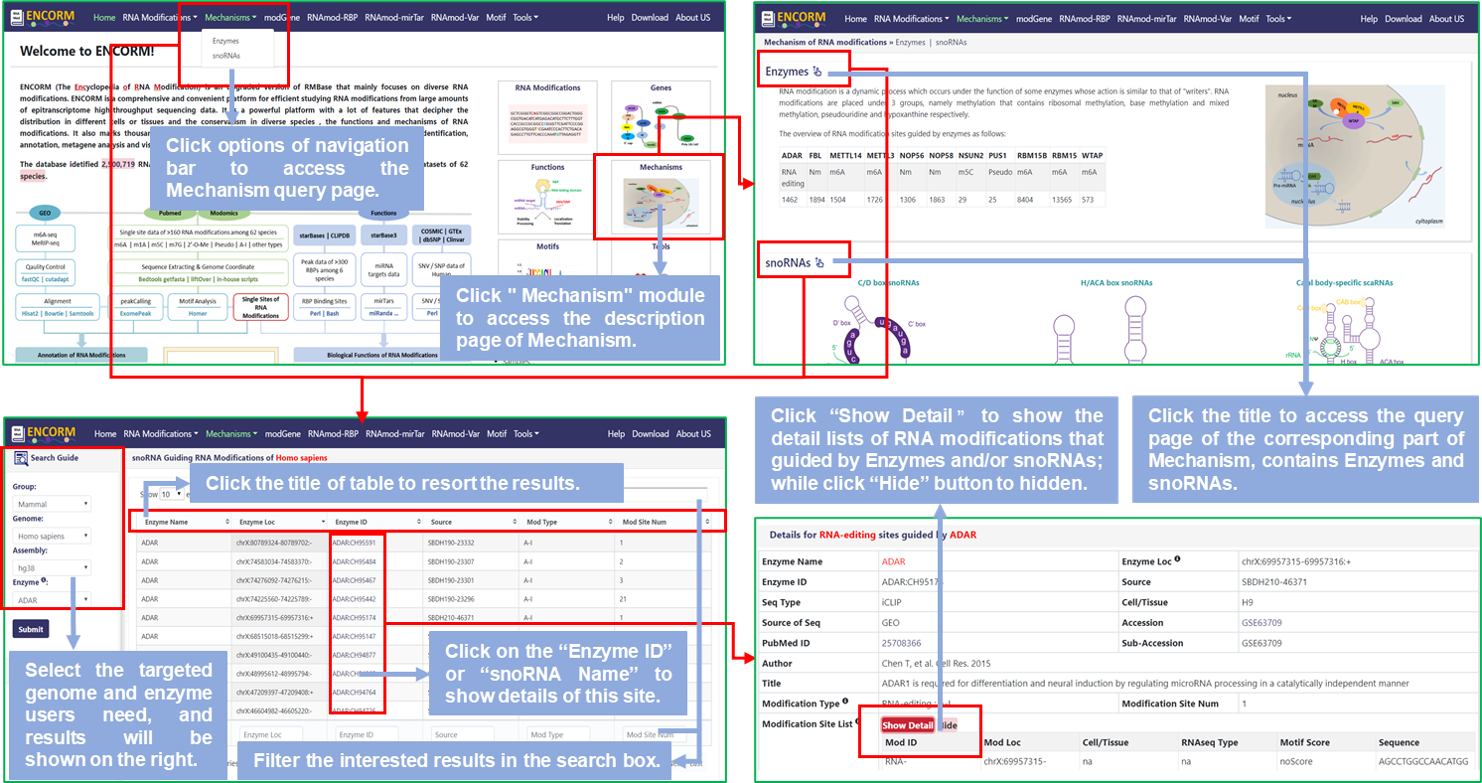
Fig.2 The workflow of "Mechanisms" module
Descriptions:
♥ Enzyme, guiding nucleotide modification of RNA, that include diverse Methylase for m6A, m5C and 2'-O-Me, Pseudouridine Synthase for Pseudo, and Adenosine Deaminase for RNA editing, respectively.
♥ The major role of snoRNA is guiding RNA modifications of snRNA or rRNA. Generally, snoRNAs are classified into box C/D and box H/ACA based on conserved motifs. Box C/D (box C: TGATGA and box D: CTGA) snoRNAs guide 2'O-methylation of target RNAs, while Box H/ACA (box H: ANANNA and box ACA: ACA) snoRNAs guide pseudouridylation.
♥ scaRNA, like SCARNA6, possess both box C/D and box H/ACA snoRNAs conserved sequence motif features, and guide snRNA modifications transcripted by RNA polymerase II.
modGene
Manuals:
modGene contains two subpages named "Genes" and "Clusters".
*** For "Genes":
Step1: Users can access the Genes query page by clicking on the "Genes" module of the home page or the "Genes" button of "modGene" options of the navigation bar to query the distribution of various RNA modifications on different genes;
Step2: Select the targeted species and its genome, default is Homo Sapiens (hg38);
Step3: Input gene name you are interested in and click the "Submit" button, then get the distribution of various RNA modifications on the gene;
Step4: Click the title of the result table to resort the results; Or input gene name or gene ID that users require in the search box to filter the results;
Step5: Click the value of "m6A Num" or other modification to display the RNA modification sites list of this type about the gene; And the details will be displayed in the detail information section that is below the query page.
*** For "Clusters":
Step1: Users can access the Clusters query page by clicking on the "Clusters" button of "modGene" options of the navigation bar to query RNA modifications clusters on different genes;
Step2: Select the targeted species and its genome, default is Homo Sapiens (hg38);
Step3: Select the type of RNA modification you are interested in and click the "Submit" button, then get multiple RNA modification clusters on different genes;
Step4: Click the title of the result table to resort the results; Or input gene name or gene ID or gene name that users require in the search box to filter the results;
Step5: Click the value of "Cluster ID" to display the RNA modification sites list of this cluster, and the information of gene in which the cluster is located.

Fig.3 The workflow of "modGene" module
Descriptions:
♥ For "Genes", the number represents the number of RNA modification site of a certain modification type on this gene. For example, the "m6A Num" represents the number of m6A modification sites in this gene, the same in other modification. If the value of "m6A Num" is 4, it means there are 4 m6A modification sites on this gene.
♥ For "Clusters", the "clusterID" is consist of the gene name and the genomic coordinate of the cluster; And the "Dentisy" represents the average distance between RNA modifications.
RNAmod-RBP
Manuals:
Step1: Users can access the RNAmod-RBP query page by clicking on the "Functions" module of the home page or the options of the navigation bar to query the relationships between RBP binding sites derived from public database and RNA modifications identified by RMBase v3.0 and stored in our databsse;
Step2: Select the targeted genome and RBP type that contains writer, reader and eraser, then get the coresponding RBPs and their relationships with RNA modifications identified by RMBase v3.0 and stored in our database, default is m6A writer of Homo Sapiens (hg38);
Step3: Click the title of the result table to resort the results; Or input RBP name or modification type that users require in the search box to filter the results;
Step4: Click the value of "RBP ID" to view details about RBP binding sites and the related RNA modification sites in the detail information section that is below the query page;
Step5: In details, click the "Show Details" button and more detailed informations of CLIP data about this RBP will be shown, while hedden if click "Hide" button.

Fig.4 The workflow of "RNAmod-RBP" module
Descriptions:
♥ The RBP Narrow Peak is the intersection peak that derived from different datasets, while the Broad Peak is merged peak.
♥ The "CLIP Support List" is detailed informations of experimental data of RNA binding protein. Among them, the "Dataset ID" is unique and uniformly numbered for datasets of CLIP data about RNA binding protein in RMBase v3.0.
♥ The RNA modification sites in the detailed list correspond to those identified in the RNA Modification module.
RNAmod-mirTar
Manuals:
Step1: Users can access the RNAmod-mirTar query page by clicking on the "Functions" module of the home page or the options of the navigation bar to query the relationships between miRNA targets that are predicted by several softwares and RNA modifications that are identified by RMBase v3.0 and stored in our database;
Step2: Select the targeted genome and the type of miRNA target, then get all miRNA targets that contain various RNA modifications, default is mRNA targeted by miRNA of Homo Sapiens (hg38);
Step3: Click the title of the result table to resort the results; Or input miRNA name/ID or modification type that users require in the search box to filter the results;
Step4: Click the "mirTar ID" to show details about this item in the detail information section that is below the query page, that include more informations like prediction softwares about miRNA;
Step5: In details, click the "Show Details" button to view the alignment infomation about miRNA with its targets and the list of RNA modifications that located in miRNA targets, while click "Hide" to hide them.

Fig.5 The workflow of "RNAmod-mirTar" module
Descriptions:
♥ miRNA targets are predicted by several softwares containing PITAID, PicTar, RNA22, TargetScan, miRanda, miRmap and microT. The "mirTar ID" is unique and uniformly numbered in RMBase v3.0, and the "miRNA Target ID" is unique and independent of targets predicted by different software.
♥ The "Alignment" presents the pairing alignment of miRNA with its target.
♥ The RNA modification sites in the detailed list correspond to those identified in the RNA Modification module.
RNAmod-Var
Manuals:
Step1: Users can access the RNAmod-Var query page by clicking on the "Functions" module of the home page or the options of the navigation bar to query the relationships between SNV or SNP sites and various RNA modifications;
Step2: Select the targeted genome and var type, containing SNV and SNP, and then get all SNV or SNP sites and their relationships with RNA modifications that are identified by RMBase v3.0 and stored in our database, default is Homo Sapiens (hg38);
Step3: Click the title of the result table to resort the results; Or input keywords, like rsID/SNVID, modID and/or disease that users require in the search box to filter the results;
Step4: Click on the "modSNP ID" or "modSNV ID" to display the deatailed informations of RNA modification site related this SNP or SNV site in the detail information section that is below the query page;
Step5: In the details section, users can click "Show Detail" button to view more details about this item, and click "Hide" button to hide it.

Fig.6 The workflow of "RNAmod-Var" module
Descriptions:
♥ SNPs and SNVs were mapped to modification regions, extended by an additional 10 nt in both the 5′- and 3′-directions for each site.
♥ The number in the "shift Pos" column represents the offset of SNP/SNV loci relative to the modification site. The range is from -10 to 10, the positive number means the SNP/SNV site appears in the downstream of RNA modification site and the negative means upstream.
♥ For SNP, the content in "Derived Source" represents the data source of the relationship between SNP and GWAS_disease/trait. 'LD_derived' means that the SNP is derived from linkage disequilibrium(LD) analysis to extract SNPs that had high LD relationship with disease-related SNPs using a threshold of r2 > 0.5. The "SNP Source" means the source that SNP site derived from.
♥ For SNV, the value of "SNV ID" includes two naming methods. Data derived from COSMIC is named with the begining of "COSV" that is the same as ID in COSMIC; while data derived from other public databases is named with the begining of "PUB" that is unique in RMBase v3.0.
modHistone
Manuals:
Step1: Users can access the modHistone query page by clicking on the "modHistone" option of the navigation bar to query the relationships between RNA modifications and histone modifications;
Step2: Select the RNA modification and the histone modification users are interested in of the targeted genome, users can get the list of datasets of histone modification, which are used to related analysis with selected RNA modification;
Step3: Click the file name of the "Distribution" column to download the text file that could be used to draw according to the users preference;
Step4: Click the button of the "Data Plot" column to view the distribution of histone peak in each 500nt upstream and downstream centered on the RNA modification sites, and user can doenload the merged image or the section of image.

Fig.7 The workflow of "modHistone" module
Descriptions:
♥ The graph provided by modHistone is for reference only. Users can download the text file to draw a high-definition image.
RMP-Cancer
Manuals:
Step1: Users can click the "RMP-Cancer" option of the navigation bar to access the page for the relationships between RNA modification proteins and cancers;
Step2: Click the "Overview" button for the datasets overview of RMPs and tumors collected in RMBase v3.0;
Step3: Click the "RMP" button and select an RMP to show its expression model, based on counts or fpkm, in diverse tumors and normal tissues;
Step4: Click the "Cancer" button and select an tumor to view the differential expression of all RMPs according to samples of this tumor, which based on counts or fpkm;
Step5: In RMP details, users can click the button for visualization of the RMP expression in diverse tumors and the corresponding normal tissues;
Step6: In Cancer details, users can click the button to download text file of the differential expression for all RMPs in any two sets of samples.

Fig.8 The workflow of "RMP-Cancer" module
Descriptions:
♥ PT: Primary Tumor; STN: Solid Tissue Normal; RT: Recurrent Tumor; M: Metastatic; AM: Additional Metastatic; ANP: Additional New Primary.
Motif
Manuals:
*** For "m6A Datasets":
Step1: Users can access the Motif query page by clicking on the "Motifs" module of the home page or the "m6A Datasets" button of "Motif" option of the navigation bar to query the datasets of m6A modification;
Step2: Select the targeted species and genome, then get the sequencing and experimental informations of all datasets of m6A modification stored in RMBase v3.0, default is Homo Sapiens (hg38);
Step3: Click the title of the result table to resort the results; Or input keywords, like pubmed ID, that users require in the search box to filter the results;
Step4: Click the Accession ID, like GSExxx or GSMxxx, to view de novo identified PWMs from transcriptome sequencing data, including motif logos of m6A modification in Motif section and metagene plot that are distribution of m6A modifications on genes in Metagene section;
Step5: In the table named "Motif Info" of Motif section, users can click "More" and/or "Similar" button to show more/similar motifs of the data; In addition, users can also click "Matrix" button to download the matrix of the motif and paste it into STAMP to check the results.
*** For "All Modifications":
Step1: Users can query the Motif of variety RNA modifications about mammals anf fungus by clicking on the "All Modifications" button of "Motif" option of the navigation bar;
Step2: Select the targeted genome and RNA modification, users can get the list of top 8 motifs, default is m6A of Homo Sapiens (hg38);
Step3: Users can click the "Matrix" button to download the motif matrix file, as well as click the "PDF" or "Recerse PDF" to download the motif logo and its corresponding logo on reverse strand.
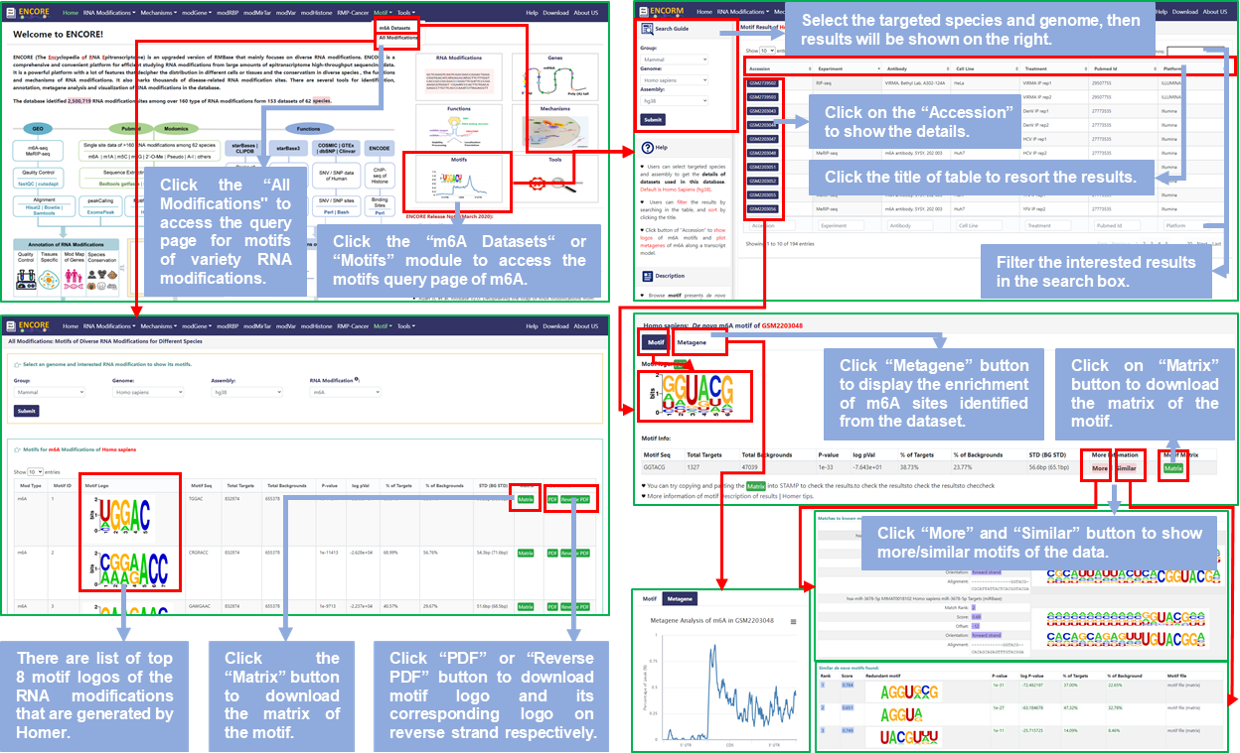
Fig.9 The workflow of "Motif" module
Descriptions:
♥ In details, motif presents de novo identified PWMs (Position Weight Matrices) from Transcriptome Sequencing Data and visualized logos of m6A motifs.
♥ In details, metagene plot display the enrichment of m6A modifications that identified from the each dataset along a transcript model.
Tools
Manuals:
Step1: Users can access the Tools page by clicking on the "Tools" module of the home page or the options of the navigation bar to analyze your data. The data contains the list of gene ID or transcript ID and peak/site data with bed6/bed6+ format;
Step2: Select the targeted species and genome, default is Homo Sapiens (hg38);
Step3: Check items that you want to analyze and the program will begin after you click the submit button;
Step4: You can directly click the result link or copy the Task ID and paste it into the search box of tools page to access the result page and view the analysis results;
Step5: In the results page, you can scroll uo and down the page to view all the graphs and tables of results. If you want to download all results, you can click the "Export" button above the graph/table or click the "Download" button on the left side of the page to download all results with the compressed file.

Fig.10 The workflow of "Tools" module
Descriptions:
♥ See the result pages for the description documents of diverse tools.